Recall the basic idea behind Monte Carlo methods in materials science
Describe how Metropolis Monte Carlo samples equilibrium configurations
Identify the main steps in a kinetic Monte Carlo algorithm
Interpret when Monte Carlo methods are useful compared with continuum and molecular dynamics approaches
Recap: two ways for modelling large-scale motion in materials
Continuum modelling / phase-field method: described by governing diffusion (Cahn-Hilliard type) equations
Example: derivation of Einstein relation of diffusivity
Discrete / event-based method: describe system as discrete steps
Example: random walk model for diffusion
Distinction:
Do we have a continuum description of properties?
Is probability easier to calculate?
Recap: comparison between scales
What is Monte Carlo method, anyway?
Monte Carlo (MC) methods describes a large set of stochastic methods to perform sampling in a (usually) high-dimensional space.
Monte Carlo is part of the Principality of Monaco renowned for casino, luxurious hotels etc 👉 statistical mechanism was historically associated with gambling!
First coined by von Neumann, Ulam, and Metropolis in Los Alamos to study neutron diffusion
MC methods work surprisingly well for distribution in high-dimensional space, coupled with Bayesian statistics
View of Monaco beach. Wikipedia
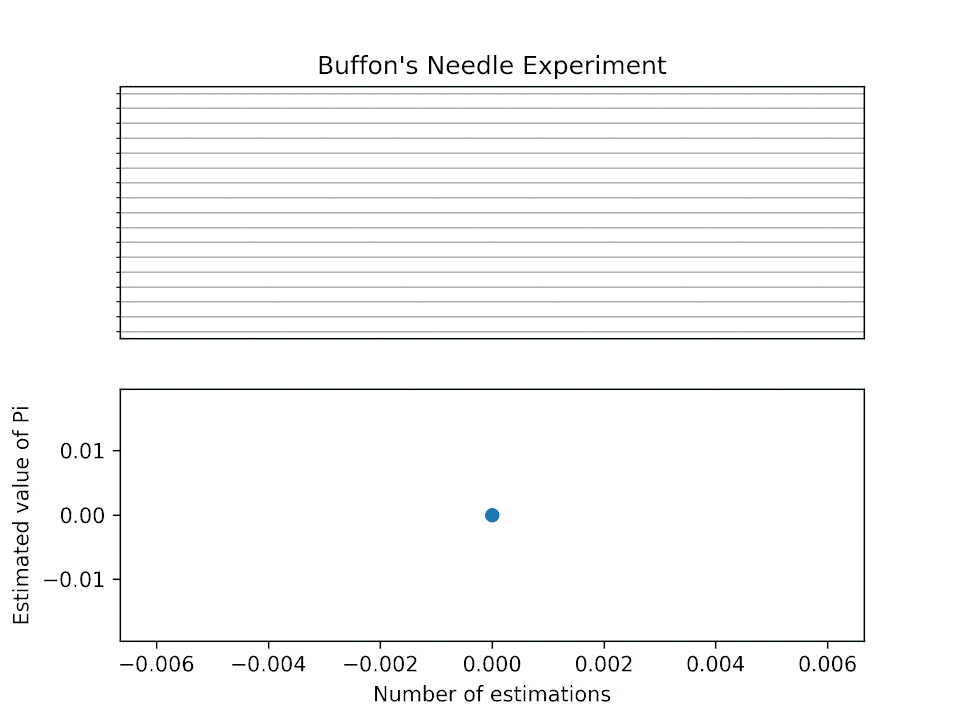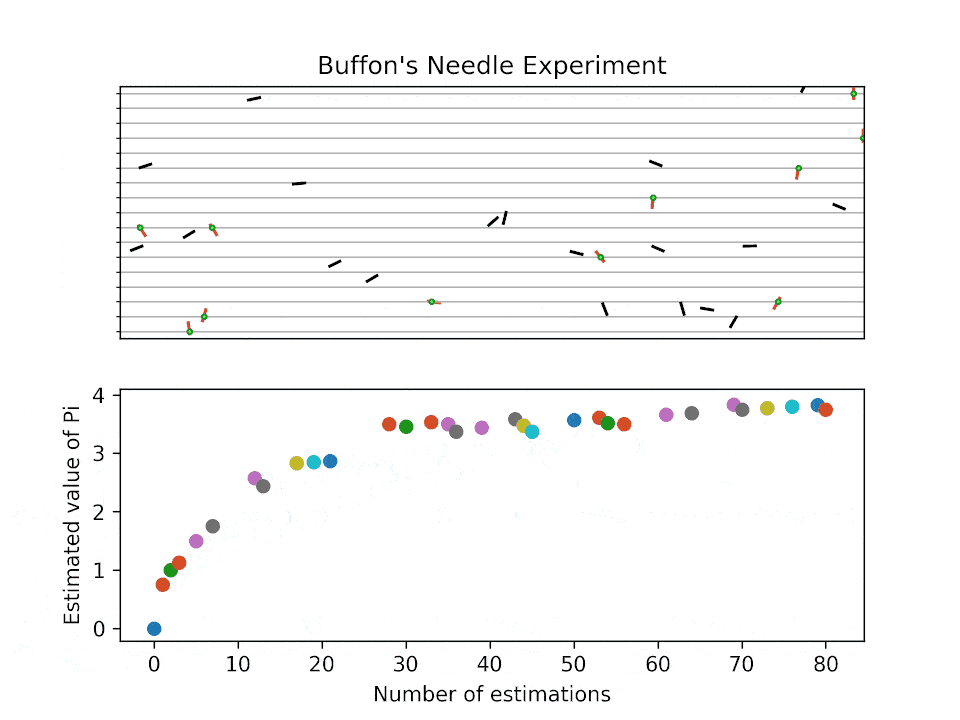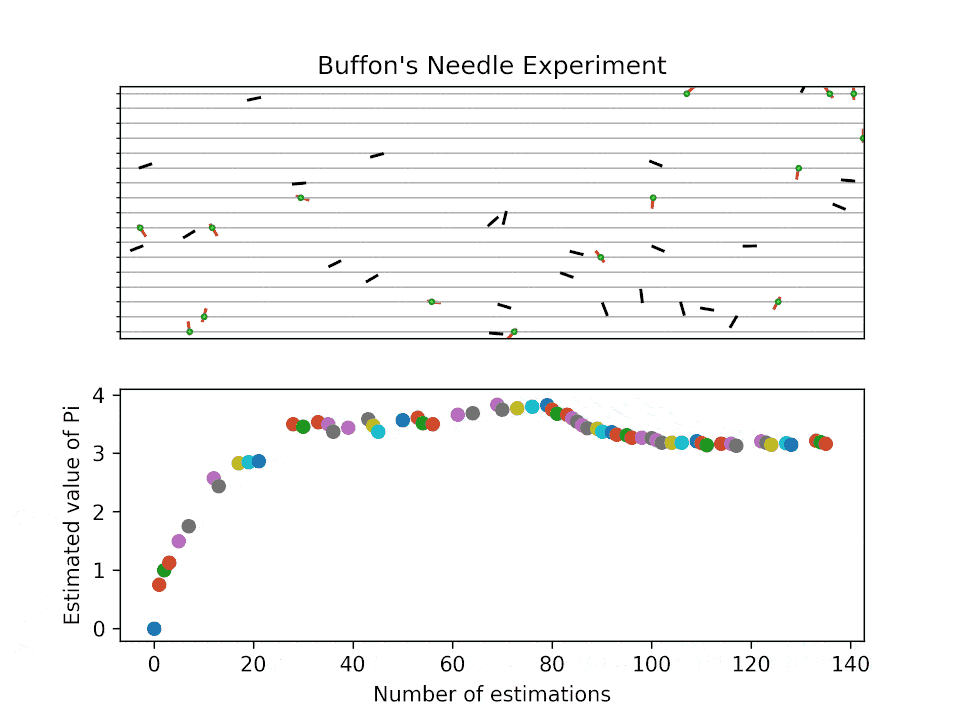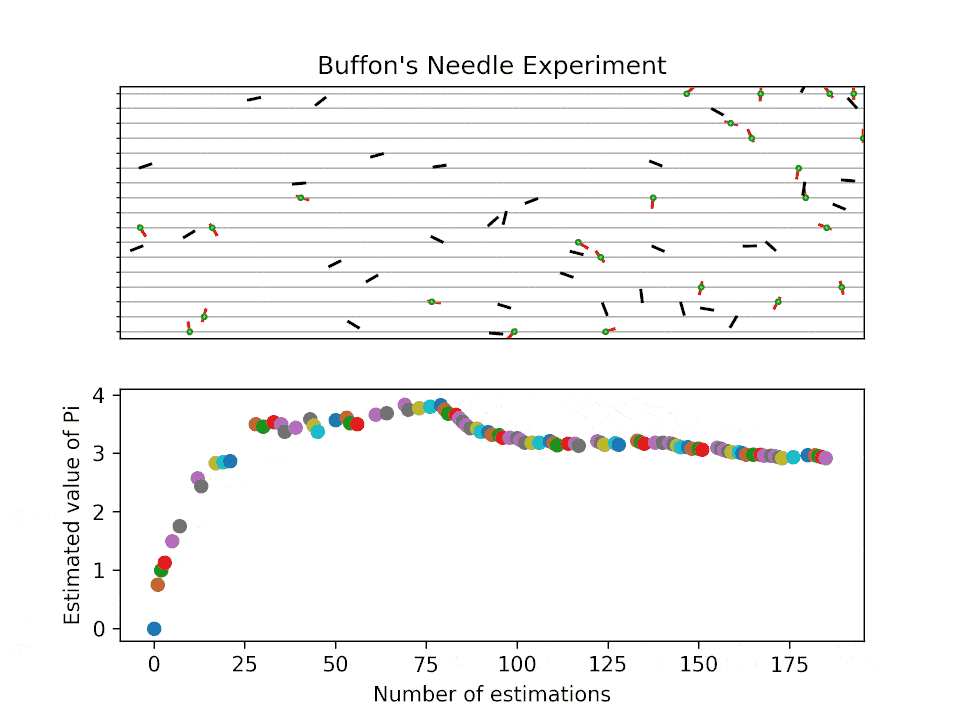
Historical background for sampling-based method: Buffon’s needle problem
Buffon’s needle problem is one of the earliest demonstrations to estimate an unknown quantity (here \(\pi\)) via sampling. The probability of a needle with length \(l\) intersecting parallel lines spaced at \(d\) is:
\[
p = \frac{2}{\pi} \frac{l}{d}
\]
How does the estimation work?
Recently proposed method for finding \(\pi\) using just coin-flip
Buffon’s needle problem: visualization
How fast do the MC sampling converge?
Can such integration be distributed over different systems?
Simulation from wikipedia
Have we already seen a Monte Carlo simulation in our previous lectures?
Yes. The vacancy-mediated diffusion process (Lecture 9 & Assignment 2) shows this approach. We know that the macroscopic diffusion must follow the Fick’s equation
\[
J_A = -\tilde{D}\nabla c_A
\]
and we can simulate the process (i.e. not knowing \(\tilde{D}\)) while only caring about the probabilities that a vacancy exchanges with a certain type of atom.
Why are the probability picture and diffusion the same thing? 👉 See answers to Assignment 2
What do Monte Carlo simulations solve?
The following are taken from Landau and Binder, A Guide to Monte Carlo Simulations in Statistical Physics, (Cambridge U. Press, Cambridge U.K., 2000).
In a Monte Carlo simulation, we attempt to follow the ‘time dependence’ of a model for which change, or growth, does not proceed in some rigorously predefined fashion (e.g., according to Newton’s equations of motion) but rather in a stochastic manner which depends on a sequence of random numbers which is generated during the simulation.
Monte Carlo method vs molecular dynamics
The Monte Carlo (MD) method and Molecular Dynamics (MD) are both simulation techniques in material science but can be easily confused.
Similarity
Use of potential energy / force field: the energy difference in MC and MD may be coming from similar or even the same potential energy equations
Molecular representation: both MC and MD may be used to study exact atomic positions (explicit atom models) or coarse-grained models (groups of atoms)
Ensemble: can be used in both cases
Difference
MD simulations follow Newton’s equation
(Usually) fixed time steps
Calculation of forces (= gradient in energy) needed
MC simulations use the probability between transitions
Can advance in varied time steps (kinetic MC)
Don’t need to calculate momenta / forces
Where does the probability come?
One main topic in the kinetics course is how to link the probability of a system to a certain energy scale. For a property \(A\) of interest, in the canonical (NVT) ensemble of system, such distribution follows the Boltzmann distribution
To estimate the expectation of \(A\) in NVT ensemble, we are required to calculate:
\[
\langle A \rangle_{NVT} = \frac{\int_\Omega A \exp(- \frac{U}{k_B T}) d\mathrm{r}}
{\int_\Omega \exp(- \frac{U}{k_B T}) d\mathrm{r}}
\]
How do we compute the integrals?
Mathematical form of Monte Carlo integration (I)
In the center of Monte Carlo methods, we are dealing with the integral of some (presummably) high-dimensional function \(f(\mathbf{x})\) on a domain \(\Omega=\{x_1, x_2, \cdots, x_n\}\), which can be transformed by another probability density function \(\rho(\mathbf{x})\)
\[\begin{align}
F &= \int_{\Omega} f(\mathbf{x}) d \mathbf{x} \\
&= \int_{\Omega} \frac{f(\mathbf{x})}{\rho(\mathbf{x})} \rho(\mathbf{x}) d \mathbf{x}
\end{align}\]
The integration problem for \(F\) now effectively becomes to find the expection for \(f / \rho\), when we draw samples \(x\) from the distribution \(\rho(x)\):
\[\begin{align}
F &= \int_{\Omega} \frac{f(\mathbf{x})}{\rho(\mathbf{x})} \rho(\mathbf{x}) d \mathbf{x} \\
&= E\left[ \frac{f(\xi)}{\rho(\xi)} \right]
\end{align}\]
When the distribution \(\rho(x)\) is uniform, then we have
\[\begin{align}
F \approx \frac{\Pi_{j=1}^{n} L_j }{N_{\text{trial}}} \sum_{i=1}^{N_{\text{trial}}} f(\xi_i)
\end{align}\]
where \(L_i\) is the domain size in dimension \(j\).
Mathematical form of Monte Carlo integration (III)
Why does the Monte Carlo method work? Compare the two ways to calculate \(F\)
\(F\) by integration \(F = \int_{\Omega} f(\mathbf{x}) d \mathbf{x}\)
Requires at least \(\text{npt}^n\) data points
Intractable in high-dimensional space
\(F\) by Monte Carlo sampling \(F \approx \frac{\Pi_{j=1}^{n} L_j }{N_{\text{trial}}} \sum_{i=1}^{N_{\text{trial}}} f(\xi_i)\)
Requiring \({N_{\text{trial}}}\) sampling steps
No exponential expansion with dimension (but may require convergence check)
Performance differ by the choice of \(\rho\)
Statistical mechanics from MC perspective
The expectation value for \(A\) in a statistical mechanics system using a uniform sampling strategy is now:
This is a powerful conclusion! Every single thermodynamic quantity in the current ensemble can be estimated by random sampling. A long-enough MC simulation will eventually reveal the property.
Is this good enough?
Problem with uniform-sampling Monte Carlo in materials simulations
The uniform-sampling MC method is not a feasible approach. There are a very large number of configurations that would be randomly generated that have effectively zero Boltzmann weight due to high-energy overlaps between the particles.
Most points in the phase space: high-energy configurations 👉 extremely low probability
Low energy configurations: physically observed states 👉 extremely low phase-space density
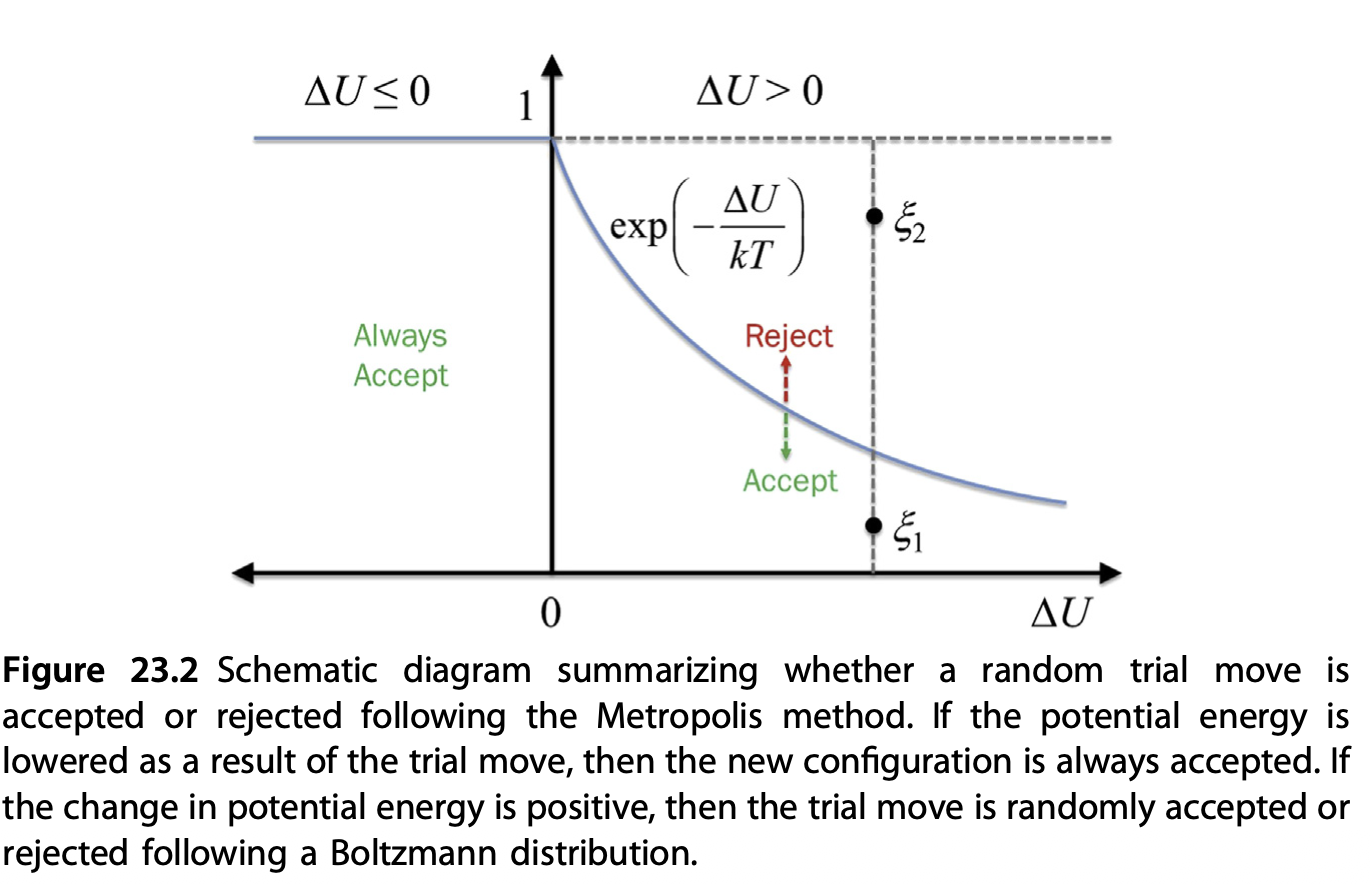
The solution: importance sampling / Metropolis-Hastings algorithm
Let’s review the integral question again. Do we really need to use a uniform probability distribution \(\rho\), in systems that has Boltzmann behavious?
\[
F = \int_{\Omega} \frac{f(\mathbf{x})}{\rho(\mathbf{x})} \rho(\mathbf{x}) d \mathbf{x}
\]
Key idea: if \(\rho(\mathbf{x})\) is close to the actual \(f(\mathbf{x})\), the observed quantity during each sampling, \(\frac{f(\mathbf{x})}{\rho(\mathbf{x})}\) has little variance
Problem: we don’t know what shape function \(f\) looks like, so can just guess.
The best guess we could have is just the Boltzmann distribution
Probability follow Boltzmann distribution \(p \propto \exp\left(-\frac{E + C N}{T}\right)\)
Scaled potential / entropy \(C = T \mu = T \partial S/\partial N\)
A modified grand canonincal Monte Carlo (GCMC) simulation: atoms can be inserted / removed to a reservoir with chemical potential \(C\)
Metropolis MC: the Potts model for polycrystalline materials
Potts model (similar to the Icing model in solid-state physics) is a widely used technique to simulate the spatial composition domains in polycrystalline materials using the Monte Carlo method.
Interested in:crystalline orientation \(q_i\) in each domain & how large the domains are
Mathematical formulation:
\[
H = \sum_{\langle i,j\rangle} J \left(1 - \delta_{q_i,q_j}\right)
\]
\(\langle i,j\rangle\): neighboring lattice sites (up to 3rd- or 4-th neighbours)
\(\delta_{q_i,q_j}=1\) if \(q_i=q_j\), otherwise \(0\): penalty for creating a grain boundary
Different neighbors increase the energy \(\Rightarrow\) grain boundary energy
Potts model simulation
In a Metropolis MC simulation using the Potts model, Monte Carlo algorithm updates
Monte Carlo in temporal domain: kinetic Monte Carlo (KMC)
From our previous examples we clearly see the (Metropolis) MC method is a powerful tool to reveal equilibrium composition, but what about systems that evolve over time?
Solution: Kinetic Monte Carlo (KMC)
Provides much longer time scale than MD
Probability comes from energy barrier / transition states
Fundamentals from the Markov Chain (MC) process
KMC in a nutshell
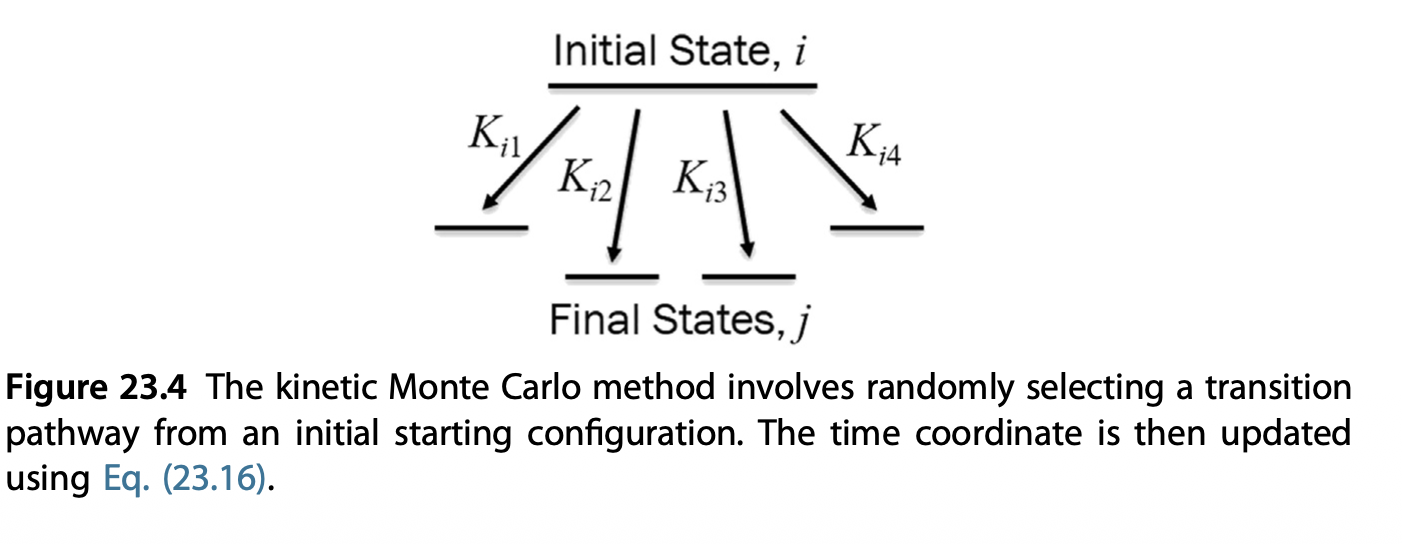
The KMC modelling is a special case of Markov-Chain Monte Carlo (MCMC), which tries to answer the question: how fast can I move from states to the other other.
From each state \(i\), there are multiple states \(j\) that it can move to
The rate of \(i \to j\) depends on the kinetic energy barrier between them
From \(i \to j\), the most probable state is picked by considering all possible paths
Time \(t\) is not explicitly used in the evolution but as a “process tracker”
KMC algorithm
We will introduce the direct method of KMC
Initialize the system at \(t=0\).
Find all possible transition paths for current state \(i\) either by:
Eigenvector-following method
Nudged-elastic band method
For each path, identify the end state \(k\)
Calculate individual rate \(k_{ij} = k_0 \exp(-\frac{E_{ij}^a}{k_B T})\) and total transition rates \(K = \sum_j k_{ij}\)
Pick a random number \(RN_1\), select the transition pathway \(i \to j_k\) that follows
\[
\sum_{j=1}^{j_k - 1} k_{ij} < RN_1 \cdot K < \sum_{j=1}^{j_k} k_{ij}
\] 6. Move the system to state \(j_k\), and repeat step 2
Time handling in KMC simulation
If time needs to be tracked instead of the simulation steps, after selecting the pathway, the system time advances by
\[
\Delta t = -\frac{1}{K} \ln RN_2
\]
where \(RN_2\) is a second independent random number.
This is because for ANY of the first-order \(i \to j\) process to happen, the time scale is \(\frac{1}{K}\), while less likely events scales exponentially.
First random number \(RN_1\): which process to happen?
Second random number \(RN_2\): how long does it take to happen?
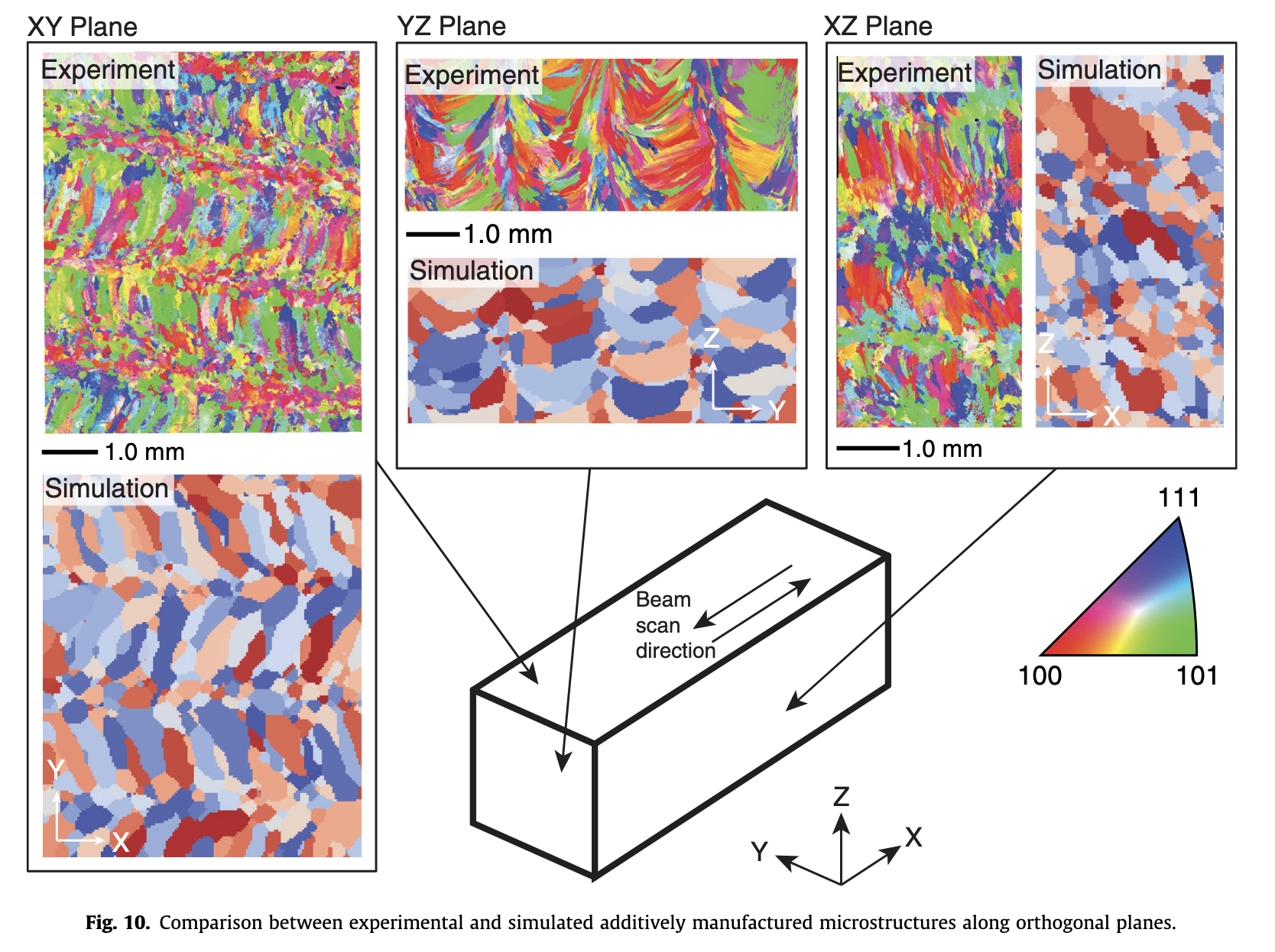
KMC simulation: additive manufacturing example
KMC simulation can handle much large length and timescales than MD.
Comput. Mater. Sci._ 135, 78–89 (2017).
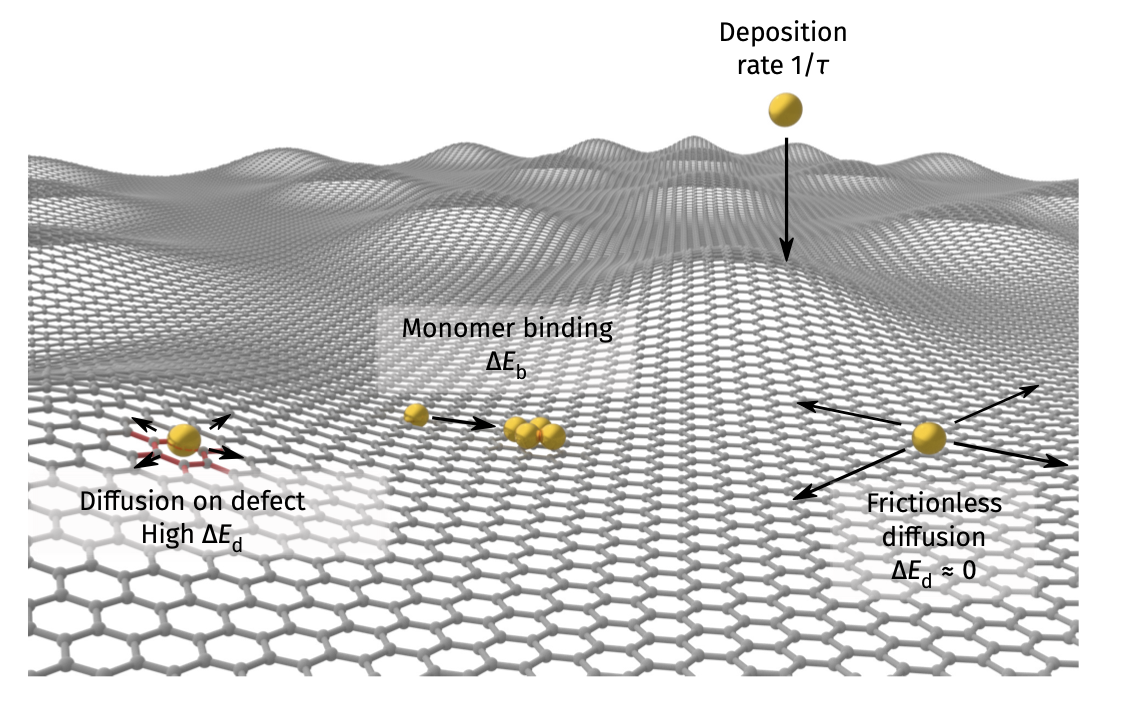
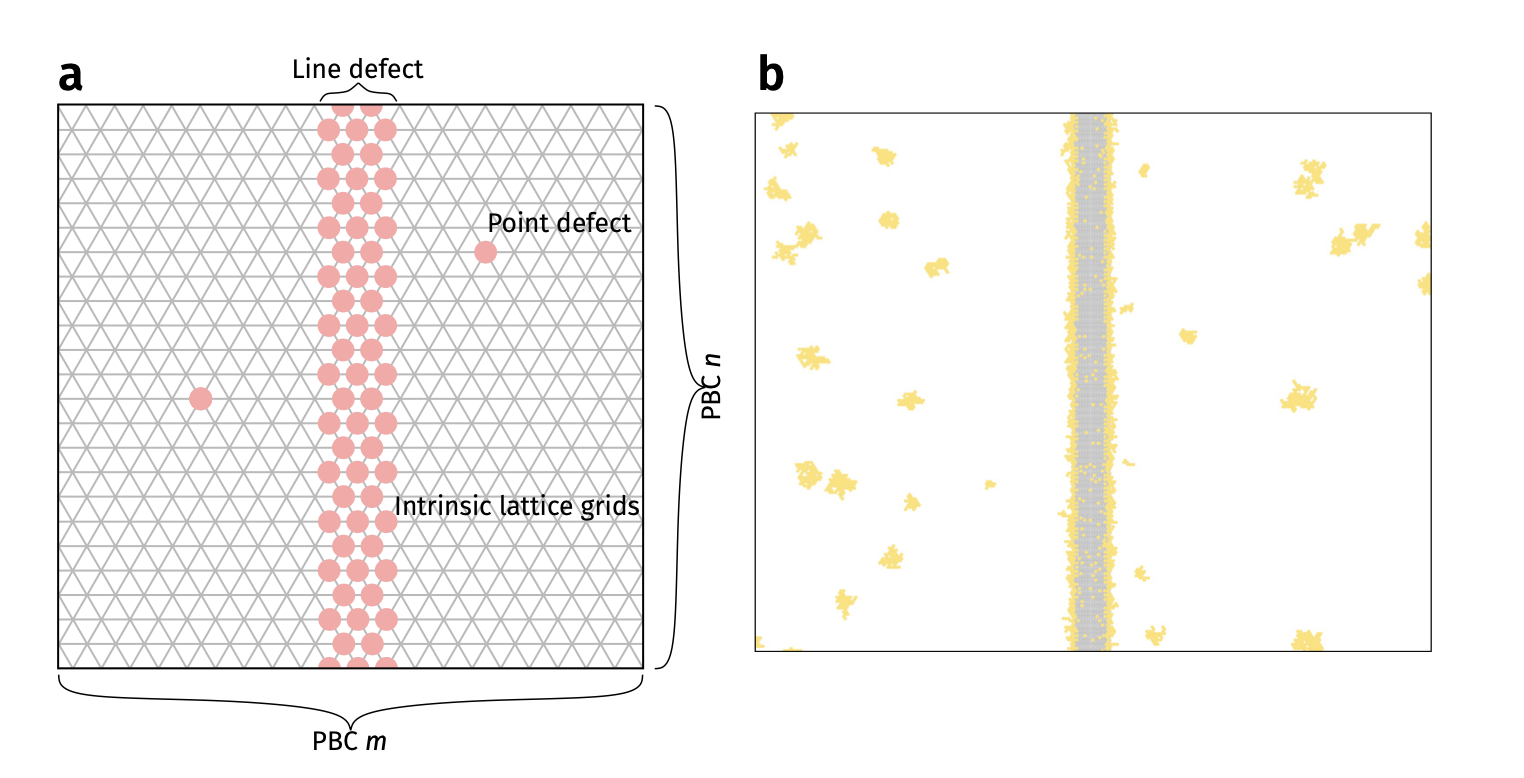
KMC example 2: deposition on graphene (previous Lecture 11)
See Vagli and Tian et al. Nat Commun 2025, 16, 7726.
Diffusivity change on free graphene surface can be probed by deposition geometry!
Deposition on Graphene - theoretical simulations
Kinetic Monte Carlo (kMC) assuming different diffusion barrier on imperfections
Faster diffusion direction –> lower density
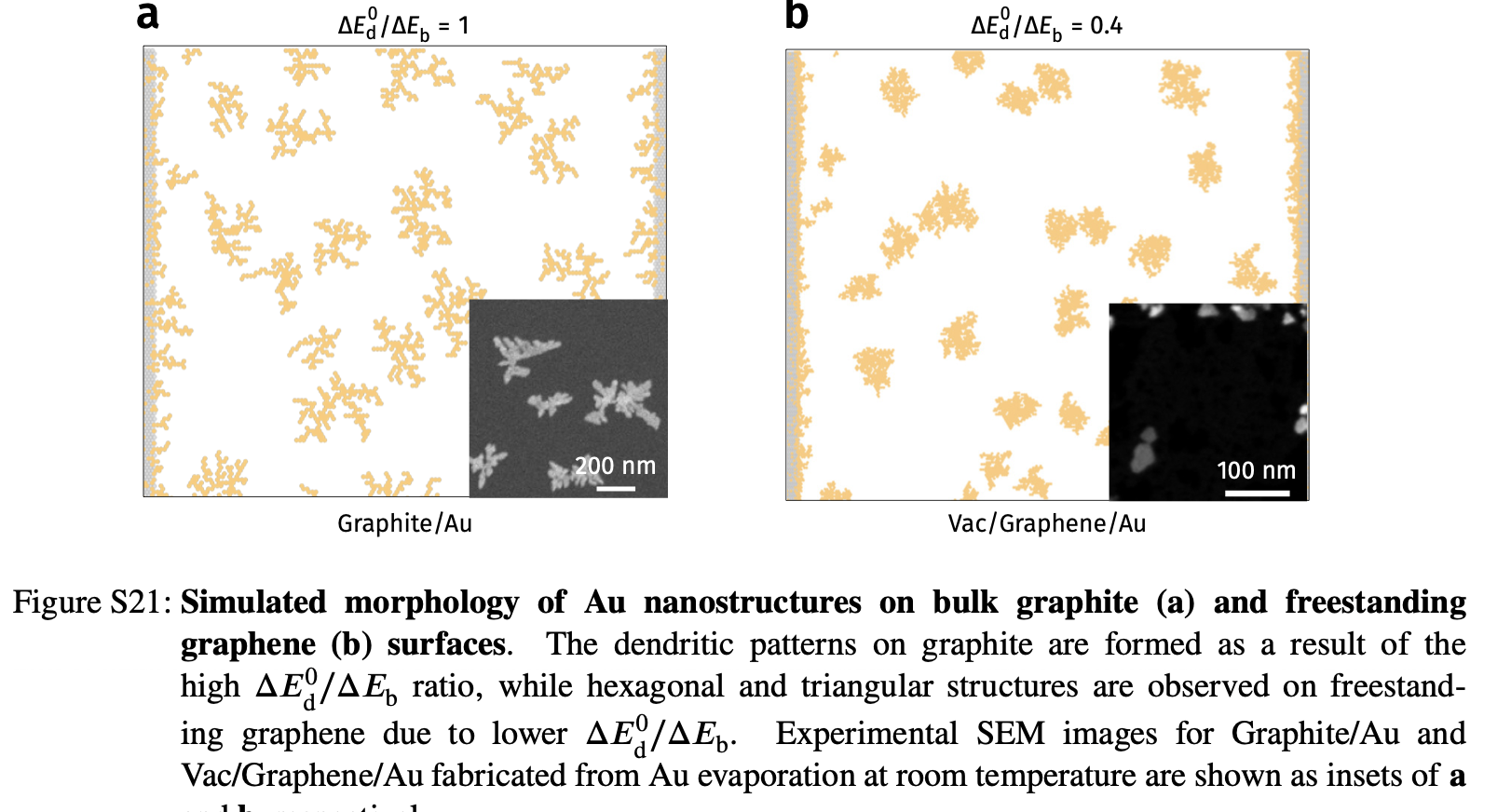
Deposition on Graphene - theoretical simulations
Kinetic Monte Carlo (kMC) assuming different diffusion barrier on imperfections
Faster diffusion direction –> lower density
Deposition on Graphene - KMC vs experiments
Summary
Monte Carlo methods use stochastic sampling to study materials problems that are difficult to solve deterministically
Metropolis Monte Carlo targets equilibrium sampling, while kinetic Monte Carlo tracks rare events over time
KMC links transition rates, pathway selection, and elapsed time in a discrete-event framework